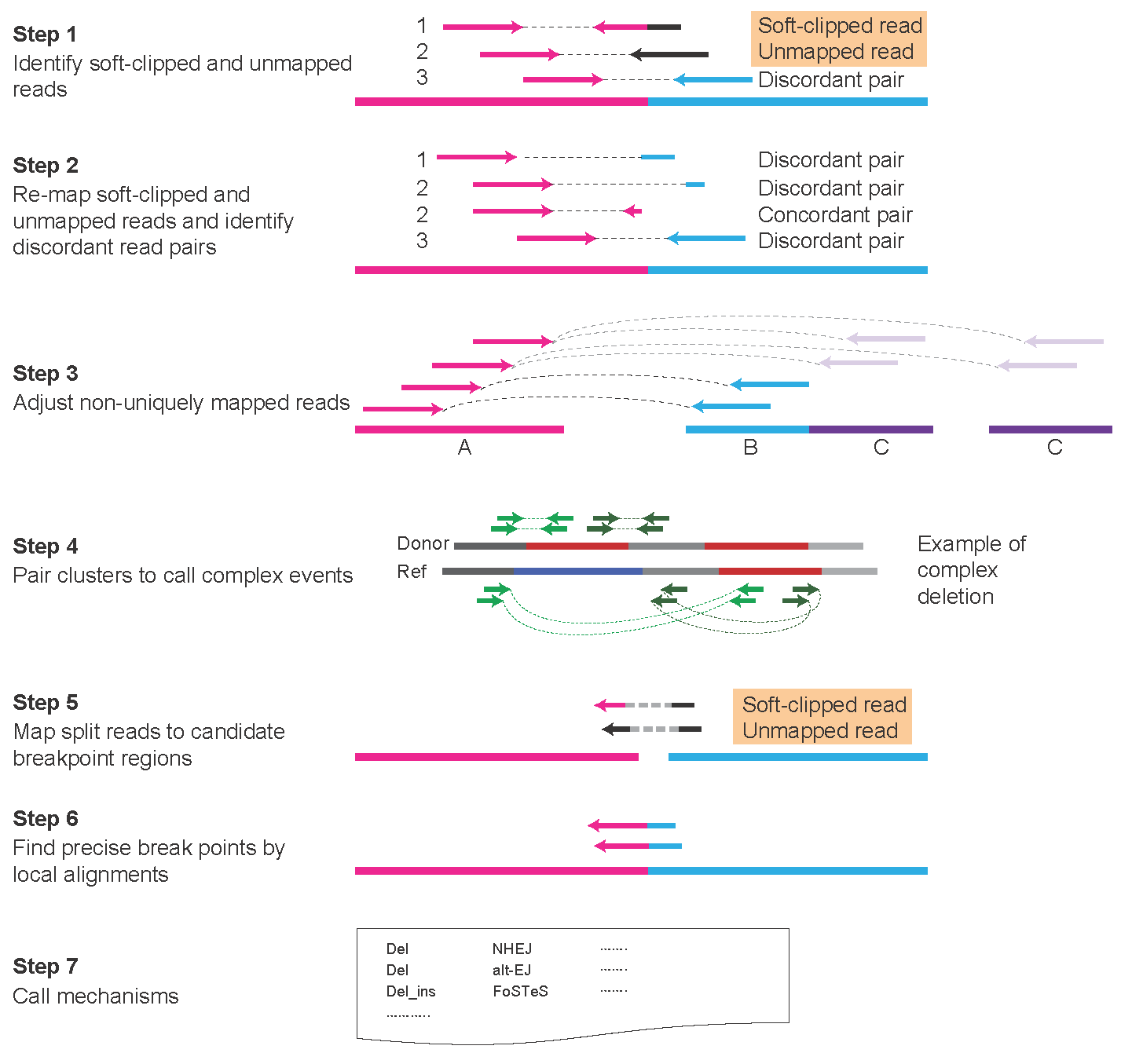
Diverse mechanisms of somatic structural variations in human cancer genomesLixing Yang1, Lovelace J. Luquette1, Nils Gehlenborg1,3, Ruibin Xi1,4, Psalm S. Haseley1,5, Chih-Heng Hsieh6, Chengsheng Zhang6, Xiaojia Ren5, Alexei Protopopov7, Lynda Chin7, Raju Kucherlapati2,5, Charles Lee6,8, Peter J. Park1,5,9,*1Center for Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA
2Department of Genetics, Harvard Medical School, Boston, MA 02115, USA
3Cancer Program, Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA
4School of Mathematical Sciences and Center for Statistical Science, Peking University, Beijing 100871, China
5Division of Genetics, Brigham and Women's Hospital, Boston, MA 02115, USA
6Department of Pathology, Brigham and Women's Hospital and Harvard Medical School, Boston, MA 02115, USA
7Department of Genomic Medicine and Institute for Applied Cancer Science, MD Anderson Center, Houston, TX 77054, USA
8Department of Medicine, Seoul National University College of Medicine, Seoul 110-799, South Korea
Houston, Texas, USA
9Informatics Program, Children's Hospital, Boston, MA 02115, USA
* To whom correspondence should be addressedCell. 153: 919-929.
.tar.gz (556KB) md5sum: 1a31a5e3b946e70d3bc592e1202ab051 .tar.gz (156MB) md5sum: b274ea762038a6d9c9cea2d8999be066
Yes! A manual is distributed with Meerkat. Before contacting us for help, please be sure to read the manual first.
If you encounter error like this:
/usr/bin/ld: gzstream.o: undefined reference to symbol 'gzclose' //lib/x86_64-linux-gnu/libz.so.1: error adding symbols: DSO missing from command line collect2: error: ld returned 1 exit status make: *** [bamreader] Error 1
Please modify the Makefile of each binary as follow:
from: ... -lbamtools -lbamtools-utils
to: ... -lbamtools -lbamtools-utils -lz
Please use the latest version of Meerkat. Meerkat requires a number of programs, modules, and references, make sure you have all of them in place and the parameters are given properly. The most likely error is some parameters are not specified correctly. If you have difficulty installing any programs, modules or references, please first contact your system admin. If you need further assistance, please contact us at ylixing@gmail.com
When you contact us, please provide the following:
Are you able to run ./bin/bamreader from command?
Are you able to run example.bam?
The output of "ls -l" for run folder.
pre.log file if it's generated.
isinfo file if it's generated.
dre.log file if it's generated.
Error message if there is any.
The refGene.txt downloaded from UCSC needs to be sorted by chromosome and coordinate by following command:
sort refGene.txt -k 3,3 -k 5,5n > refGene_sorted.txt